SeedFold: Scaling Biomolecular Structure Prediction

What is SeedFold: Scaling Biomolecular Structure Prediction
SeedFold is a new model that predicts 3D shapes of proteins and other biomolecules. It is built to be bigger, faster, and more accurate, so scientists can get good structure guesses for many kinds of tasks.

Two versions are offered. SeedFold is the flagship 923M-parameter model. SeedFold-Linear is a lighter version that runs more efficiently while keeping strong accuracy.
SeedFold: Scaling Biomolecular Structure Prediction Overview
Here is a quick look at the project, all in one place.
| Key | Value |
|---|---|
| Type | Research model and toolkit for biomolecular structure prediction |
| Purpose | Predict structures for proteins and complexes (protein–protein, antibody–antigen, protein–ligand, protein–RNA/DNA) |
| Main Models | SeedFold (923M params), SeedFold-Linear (efficient variant) |
| Training Data | 26.5M samples built by distilling AlphaFold2 outputs; sources include PDB, AFDB, Mgnify |
| Scaling Strategy | Grow model width (Pairformer 128 → 256 → 384 → 512) for higher capacity |
| Key Techniques | Linear Triangular Attention (O(n³) → O(n²)), Additive & Gated variants, optimized Triton kernels |
| Benchmark | #1 on FoldBench across many protein-related tasks |
| Notable Gains | +5.31% on antibody–antigen vs AlphaFold3; +2.99% on protein–RNA vs AlphaFold3 |
| Who Built It | ByteDance Seed team |
| Website | https://seedfold.github.io/ |
For a quick primer written in plain words, see our short explainer here: SeedFold overview on our site.
SeedFold: Scaling Biomolecular Structure Prediction Key Features
- Two model choices:
- SeedFold: the 923M-parameter flagship for top accuracy.
- SeedFold-Linear: a memory-friendly variant that keeps accuracy high.
- Strong results across tasks:
- Monomers, protein–protein, antibody–antigen, protein–ligand, protein–RNA, and protein–DNA.
- Three pillars that make it work well:
- Model scaling by width: Pairformer width grows from 128 to 512 to carry more information.
- Linear Triangular Attention: lowers compute from O(n³) to O(n²), with Additive and Gated forms.
- Large-scale distillation: 26.5M training samples from PDB, AFDB, and Mgnify.
- 2–3× memory savings thanks to the linear attention design and optimized Triton GPU kernels.
- Built to be stable at large width with special training fixes.
If you want the backstory on the team behind this work, check our short profile: ByteDance research hub.
SeedFold: Scaling Biomolecular Structure Prediction Use Cases
- Single protein structure prediction for research, screening, and annotation.
- Complex prediction, including protein–protein and antibody–antigen, to study binding and function.
- Protein–ligand docking to inform early-stage drug discovery.
- Protein–RNA and protein–DNA interactions to help with regulation studies and tool design.
- Large-scale runs where memory and speed matter, using SeedFold-Linear for efficiency.
Performance & Showcases
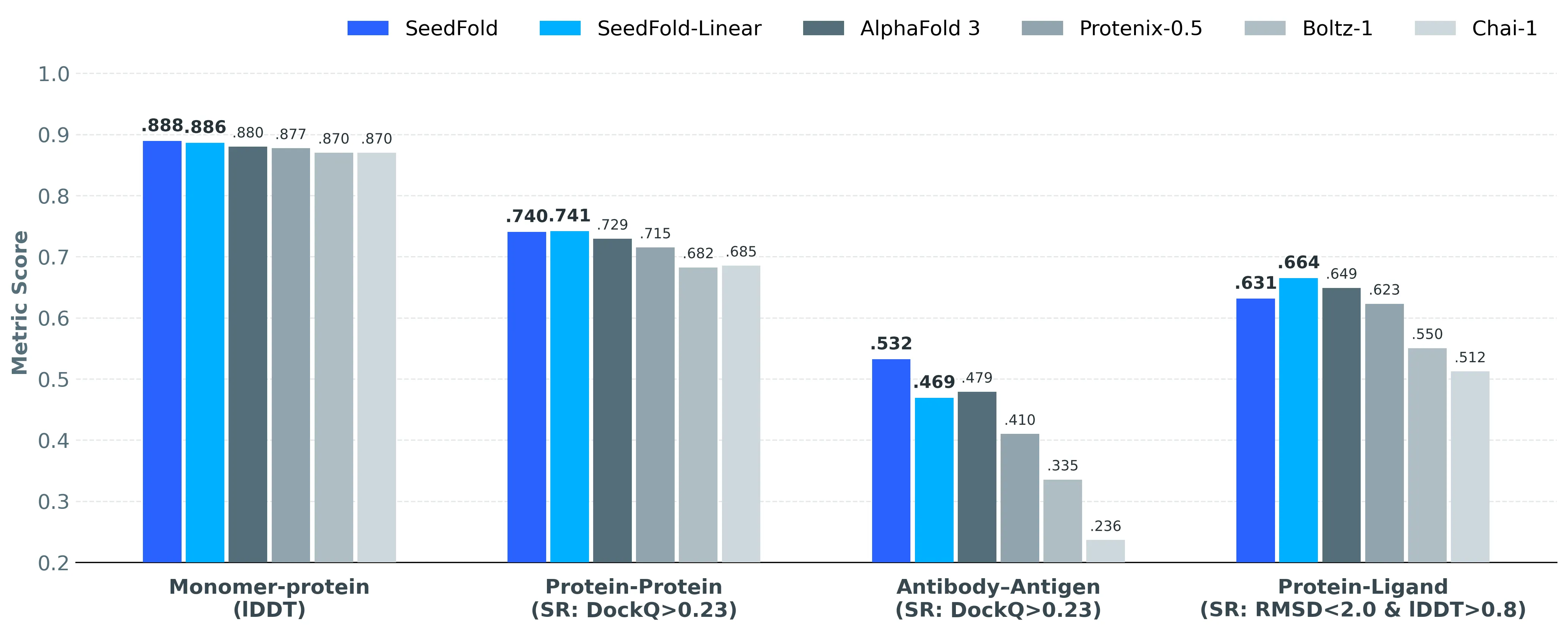
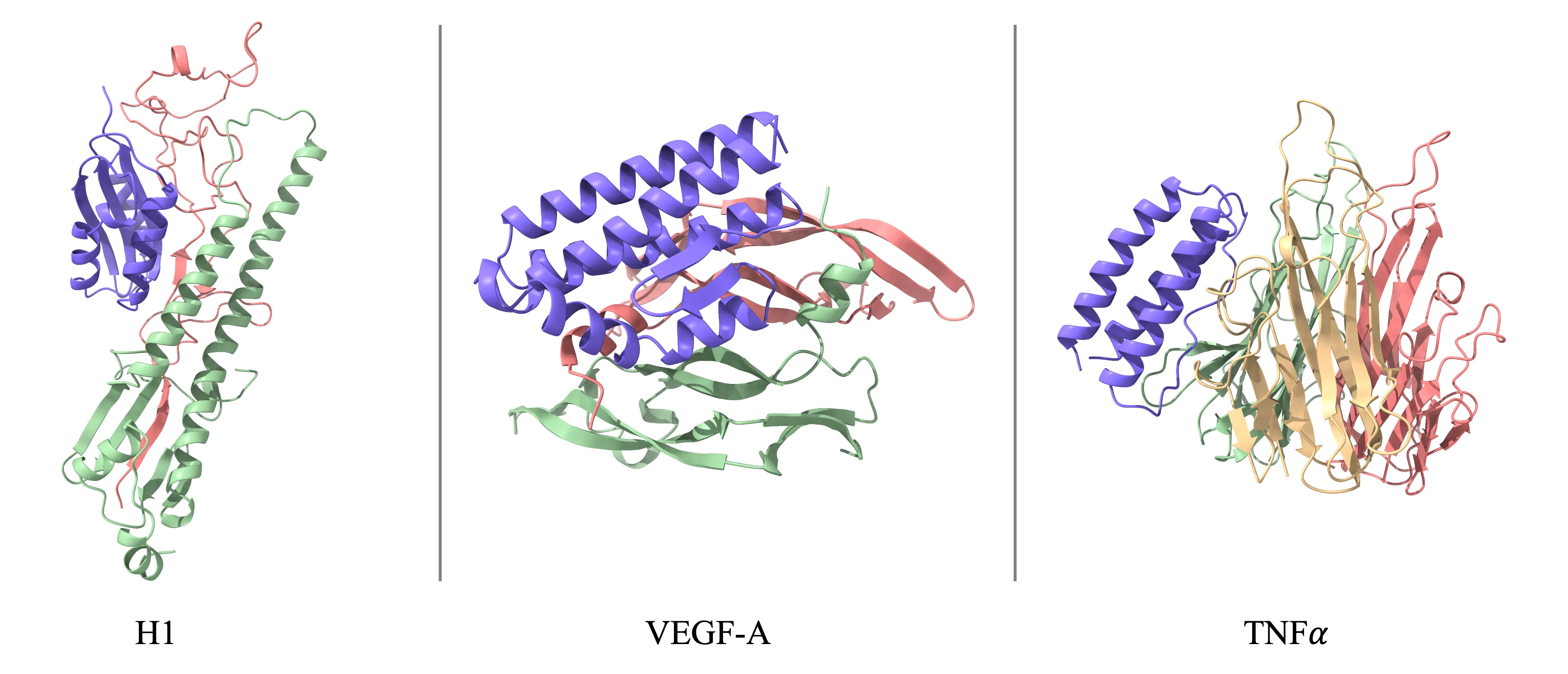
SeedFold ranks #1 on the FoldBench benchmark across many protein-related tasks. It shines on antibody–antigen (+5.31% vs AlphaFold3), protein–ligand (+1.58% in success rate vs AlphaFold3), and protein–RNA (+2.99% vs AlphaFold3). For monomers, SeedFold reaches an lDDT of 0.8889.
On protein–protein tasks, SeedFold and SeedFold-Linear both score above 74% success rate by DockQ ≥ 0.23, topping AlphaFold3 at 72.93%. Protein–DNA results are competitive, with SeedFold-Linear hitting 76.00%. Full task-by-task scores are shared on the project site.
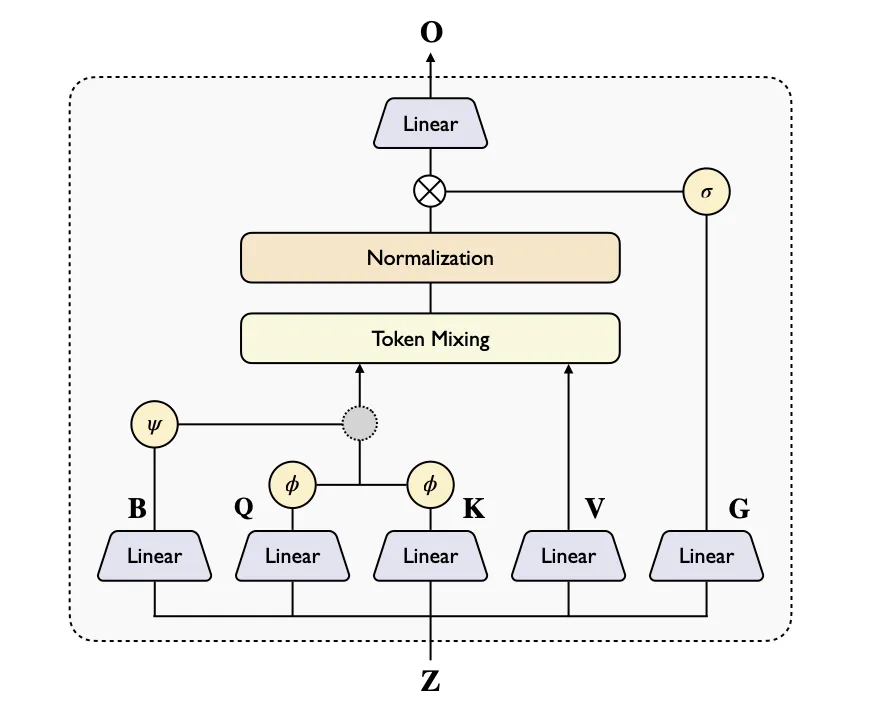
How SeedFold Works (In Simple Terms)
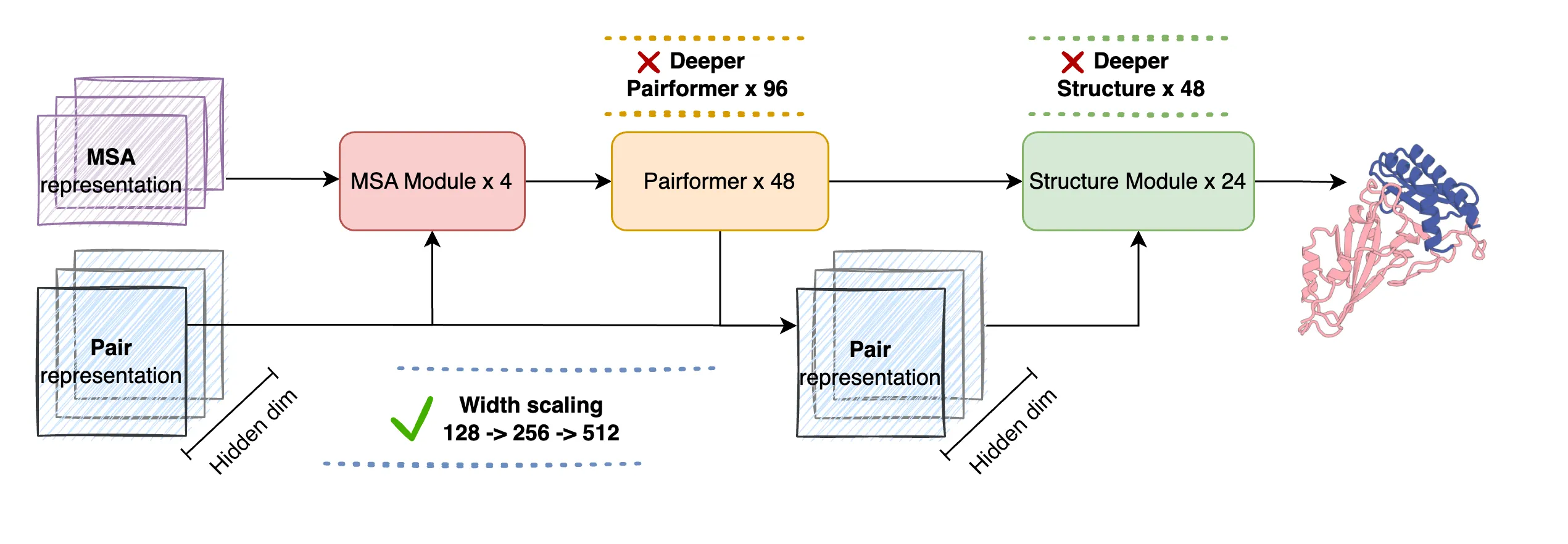
SeedFold follows a three-part flow.
First, the MSA module reads related sequences from many species and picks up patterns that hint at structure. Next, the Pairformer module builds pairwise relationships between amino acids; making this part wider gives the model room to learn richer contacts. Last, the structure module produces all-atom 3D coordinates using a diffusion-style process.
The Technology Behind It
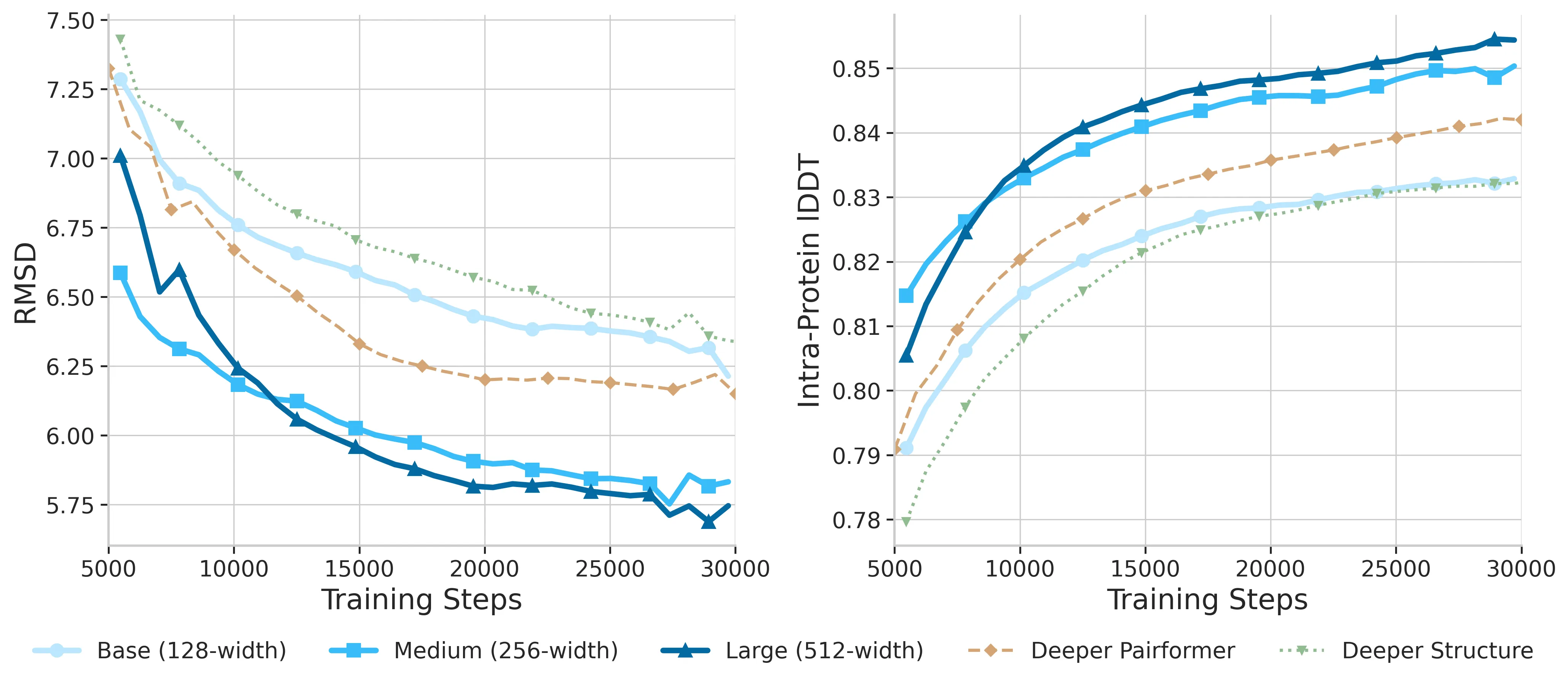
Width beats depth here. The team found that making the Pairformer wider (from 128 to 512) helps more than making the network deeper, because pair features are a key bottleneck for structure quality.
Standard triangular attention is heavy to compute. To fix this, SeedFold uses Linear Triangular Attention that scales as O(n²) instead of O(n³), with Additive and Gated versions. This brings 2–3× lower memory while keeping accuracy high; Gated works especially well on DNA/RNA jobs.
Why The Data Matters
A big part of the lift comes from data. The team built a 26.5M-sample set by distilling outputs from AlphaFold2, which is 147× larger than only using experimental structures. Sources include PDB, AFDB, and Mgnify, giving wide sequence coverage and fewer blind spots.
This scale helps the model learn many interaction types, not just the common ones. As a result, SeedFold holds up across proteins, ligands, and nucleic acids.
Related: SeedProteo for Protein Binder Design
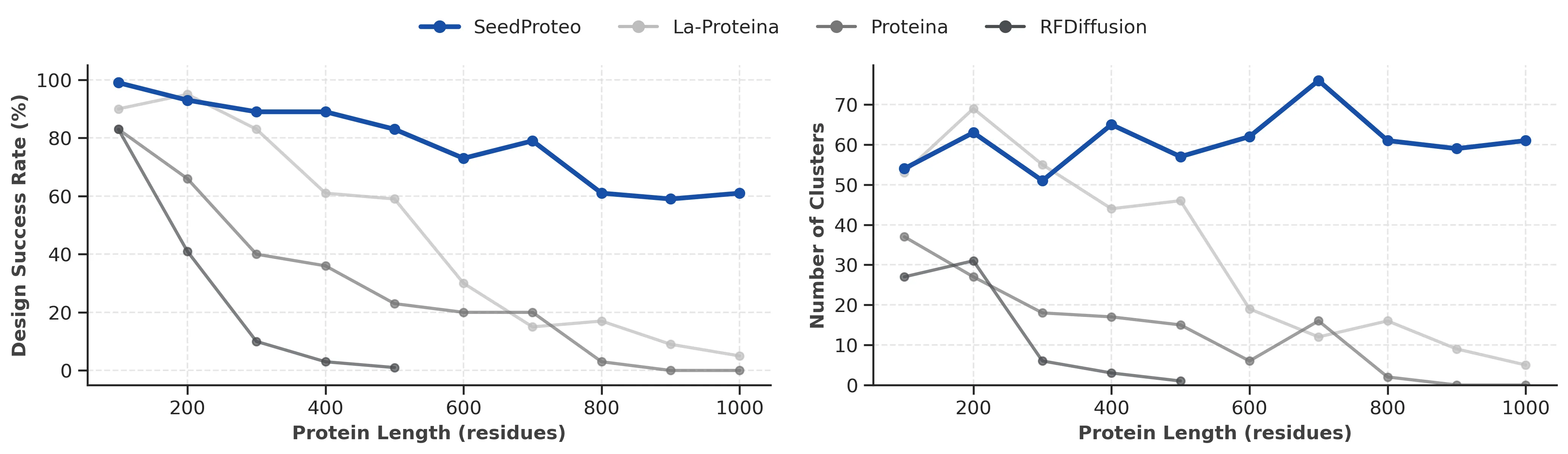
There is a sibling project named SeedProteo that designs protein binders from scratch. It borrows the folding ideas and applies a diffusion-based method to generate all-atom designs, then uses a Markov Random Field to improve sequences for global consistency.
SeedProteo reports higher success rates than open-source baselines on 10 benchmark targets. It is a clue that the same core ideas can also help design, not just predict.
Getting Started (No Code Needed)
- Visit the project site: https://seedfold.github.io/.
- Click “Try SeedFold Now” to explore the online demo and example results.
- Check “View Results” for FoldBench scores and side-by-side comparisons.
For more guides and roundups across topics, visit our home page: Omnihuman 1.Com.
Who Should Use SeedFold
- Biologists who want quick, strong structure predictions for new proteins.
- Drug discovery teams that need protein–ligand and complex modeling at scale.
- Students and labs looking for a practical tool with strong results on standard tests.
- Developers who want an efficient variant (SeedFold-Linear) to save memory on big jobs.
FAQ
What makes SeedFold different?
It scales the pairwise part of the model to be much wider, which carries more contact information. It also uses a lighter attention method to keep compute and memory in check. This balance leads to strong results across many tasks.
How is SeedFold-Linear different from SeedFold?
SeedFold-Linear swaps in Linear Triangular Attention. This lowers memory and time cost while keeping accuracy close to SeedFold. It is a smart pick for large or batched runs.
How big is the model?
The largest SeedFold has 923 million parameters. The family also includes smaller widths at 432M and 533M. Wider models showed better accuracy in tests.
What data was used to train it?
Training used 26.5 million samples built via distillation from AlphaFold2 outputs. Sources include PDB, AFDB, and Mgnify. This broad set helps performance on many structure tasks.
How does it compare to AlphaFold3?
On FoldBench, SeedFold leads on many key tasks. Gains include +5.31% on antibody–antigen, +1.58% on protein–ligand, and +2.99% on protein–RNA. Results for protein–DNA are competitive as well.
Can I try it without installing anything?
Yes. Visit the project site and use “Try SeedFold Now.” You can view examples and scores online.
Image source: SeedFold: Scaling Biomolecular Structure Prediction